InfoUpload
Upload unaligned or aligned reads from scRNA-Seq data.
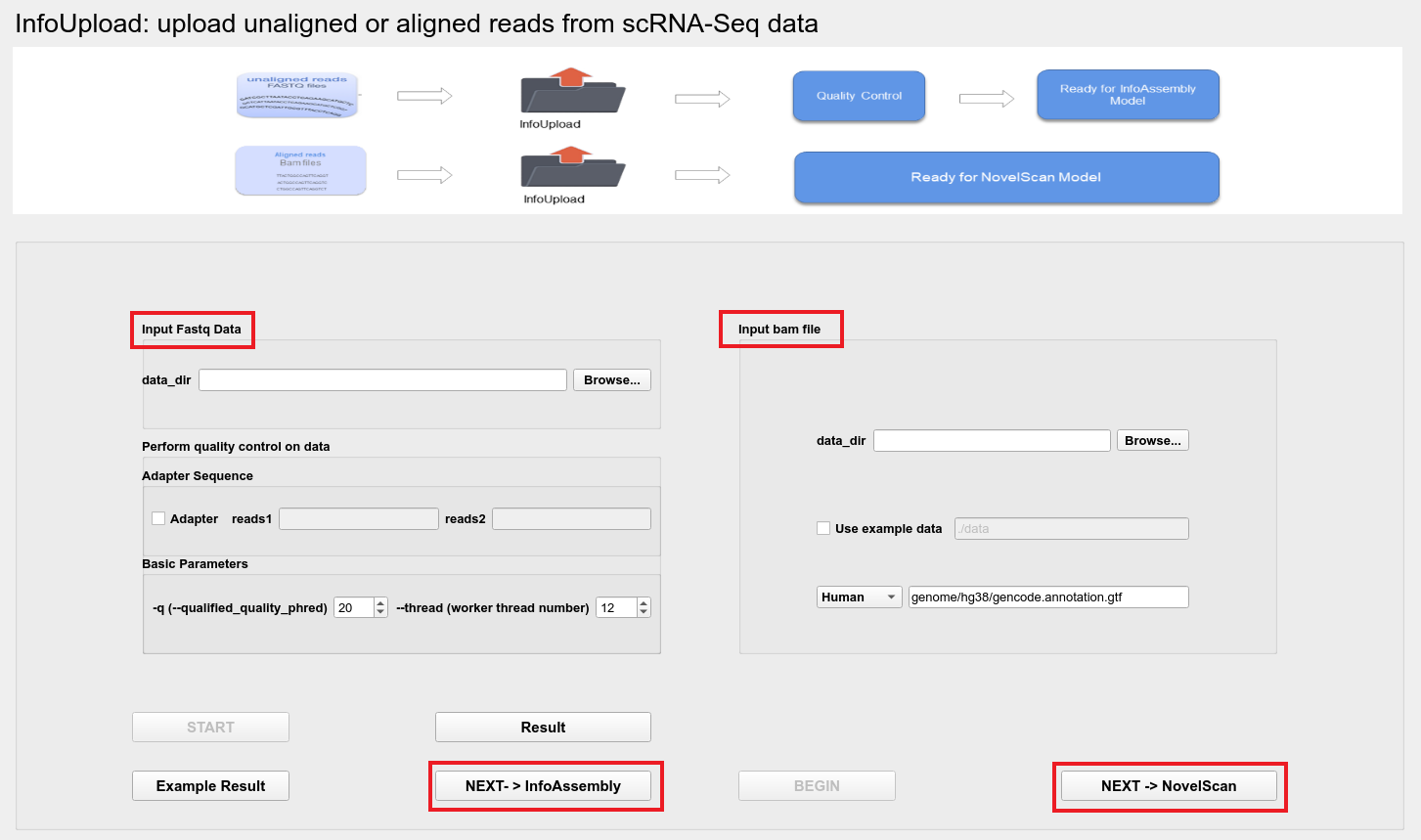
Users can choose to upload Fastq files or BAM files:
Input fastq files
The file needs to be in fastq.gz format, users can select the directory where the data is located through the Browse button;
Perform quality control on data
After uploading the data, the data will be automatically controlled for quality; if the Adapter sequence is provided, the user can enter the adapter sequence in reads1, reads2; if the adapter sequence not provided, the adapter will be automatically detected for quality control.
Basic Parameters
-q (--qualified_quality_phred)- The quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified. (int [=15]).--thread- The worker thread number.
Then the user can run the program by clicking START.
After the program runs, the user can view the results of the quality control by clicking the Result button.
Quality control results
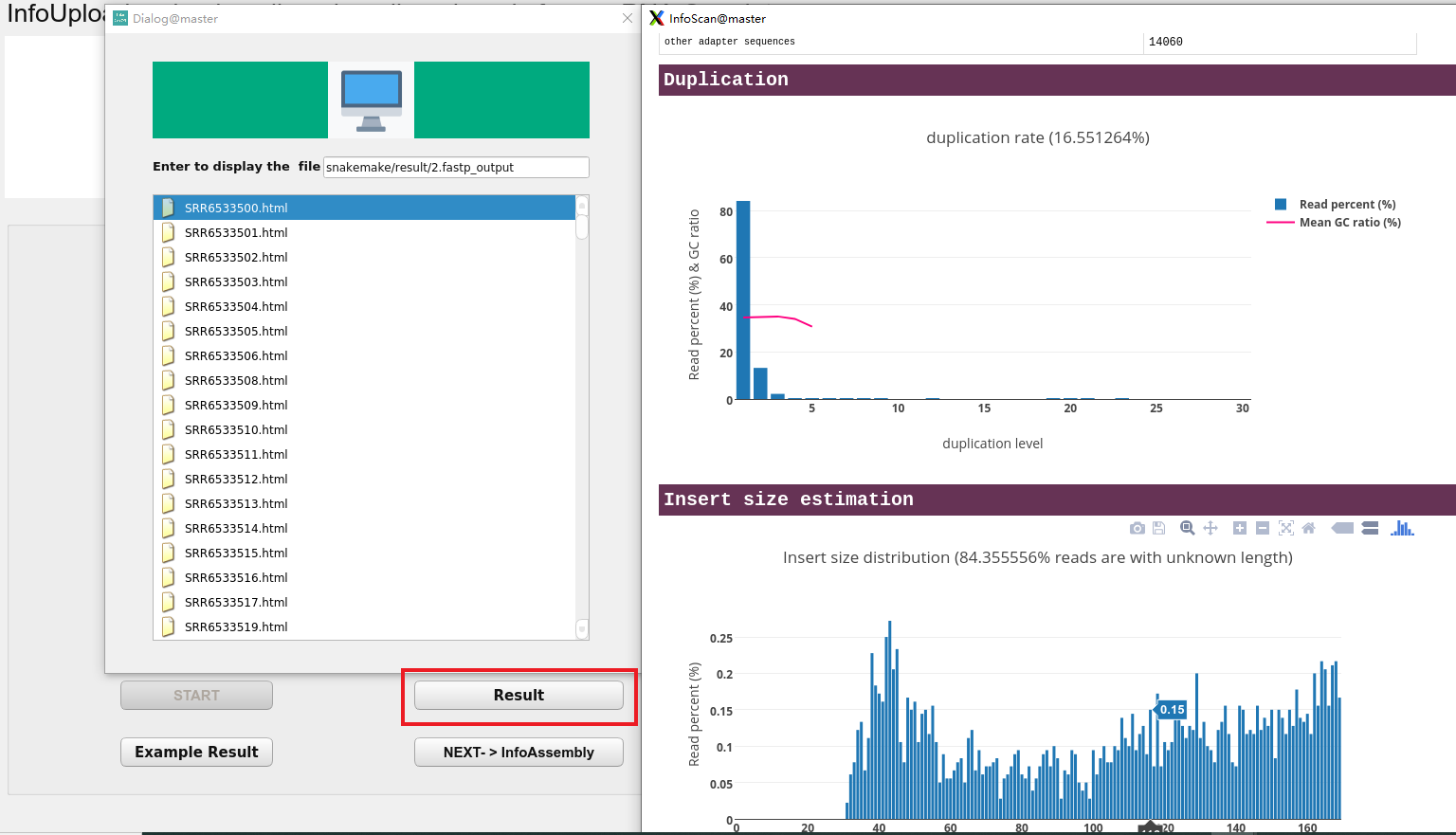
The user presses the Enter Key to display the results of the quality control and double-click the file to view the quality of the reads in the upload files.
Then, by clicking the NEXT-> Transcript Alignment button, the user can reach the InfoAssembly interface.
Input bam files
If upload bam files, the program will automatically assemble and merge transcripts.
The file needs to be in bam format, users can select the directory where the data is located through the Browse button;
Upload the example data by checking the Use example data;
Then the user can run the program by clicking BEGIN.
By clicking the NEXT-> NovelScan button, the user can reach the NovelScan interface.
Note
If the user uploads bam files, InfoAssembly will be skipped and NovelScan will proceed directly.